*Swati ** Ankit Gupta*** Ramesh Aggarwal
*Senior resident, ***Professor, Lady Hardinge Medical College, New Delhi
**Consultant, Department of Neurology, Batra hospital, New Delhi
ABSTRACT
The Neuromyelitis optica spectrum disorder (NMOSD) is an aggressive central nervous system inflammatory demyelinating disease whose diagnosis is easily missed or delayed because of its varied clinical presentations. Catatonia is an infrequent presentation of NMOSD and can present as an initial sign. Catatonia in a patient with NMOSD might be caused by autoimmune-related antibodies that attack the thalamus, hypothalamus and brainstem. Awareness of this pathophysiologic mechanism will help clinicians in the early diagnosis of NMOSD. We report a case of suspected seronegative NMOSD with catatonia as the initial presentation.
INTRODUCTION
Neuromyelitis optica (NMO)previously referred to as Devic disease, was initially described by Eugène Devic and Fernand Gault in 1894 in a case series of 16 patients. [1] At that time, the disorder was thought to be acute in onset and monophasic. Traditionally NMO was thought to have limited intracranial manifestations. Over the past decade, however, a far wider range of manifestations have been recognized as belonging together and thus the term Neuromyelitis Optica spectrum disorder (NMOSD) has been proposed to encompass them all. [2]
In approximately 70% (sensitivity of 70-90%; specificity of 90%) of patients with established NMOSD, a specific immunoglobulin can be isolated named anti-aquaporin 4 IgG (anti-AQP4-IgG) which targets a transmembrane water channel (aquaporin-4) present on astrocyte foot processes abutting the limiting membrane [3,4]. This accounts for some of the predilection for the circumventricular organs (e.g. periaqueductal grey matter) which are particularly rich in aquaporin-4 [4].
For many years, it was considered to be a subtype of multiple sclerosis, until the discovery of anti-AQP4 antibodies at which time it was moved into its own disease category. However, it is now evident that a significant proportion of patients with clinical NMOSD do not have the anti-AQP4 antibody and that the presentation can be more heterogeneous. One of the rare presentations of NMOSD is as catatonia.
Catatonia, a neuropsychiatric syndrome characterized by abnormal movements, behaviour, and withdrawal; is a condition that is most often seen in mood disorders but can also be seen in psychotic, medical, neurologic, and other disorders.[5]Studies have suggested that a connected pathway between the cortex, basal ganglia, and thalamus results in catatonic symptoms. DSM-V gives 12 categories for symptoms that can lead to a diagnosis of catatonia. These symptoms include stupor, catalepsy, waxy flexibility, mutism, negativism, posturing, mannerisms, stereotypy, agitation not influenced by external stimuli, grimacing, echolalia, and echopraxia. At least three of these symptoms must be present for the diagnosis of catatonia. [6]
We are describing a case of suspected NMOSD with catatonia as initial presentation.
CASE REPORT
A 42-year-old female presented acutely with headache, decreased mentation and somnolence, paraesthesia in bilateral lower limbs, decreased verbal output and generalized rigidity. She was drowsy but answering to questions on vigorous stimulation. She had history of low-grade fever 2 days ago. She had history of abnormal behaviour that include mumbling, wearing clothes inside out and tried to drink bread and biscuit 6 months ago following one episode of undocumented febrile illness. At that time, her abnormal behaviour lasted for a day and resolved with some medication. She had no allergies and denied taking any home medications. Her history was negative for smoking tobacco, drinking alcohol, or drug abuse. Her travel history was insignificant.
On examination, she was mute, had catalepsy, waxy flexibility, negativism and grimacing. The rest of the systemic examination was unremarkable. Psychiatric consultation was taken in view of abnormal behaviour and a diagnosis of catatonia was kept. She was admitted in medicine department and started treatment with provisional diagnosis of acute viral encephalitis vs autoimmune encephalitis. Her fundus examination did not show papilledema and lumbar puncture was done which revealed raised protein. She was started on empirical treatment of viral encephalitis.
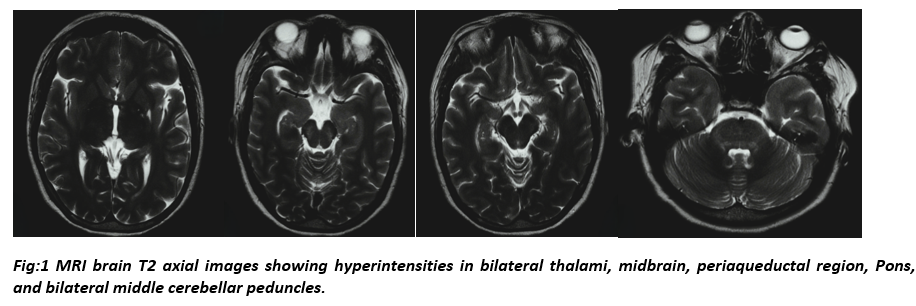
Non contrast CT head did not reveal any abnormality. She did not show any improvement for next 2 days. MRI Brain with screening whole spine obtained during this time demonstrated T2/FLAIR hyperintensities in bilateral thalami, midbrain, pons, middle cerebellar peduncles and periaqueductal grey matter (figure 1). MRI brain also demonstrated bright shinny lesion of CSF signal intensity in left insular cortex suggestive of old insult. Autoimmune encephalitis panel, anti-nuclear antigen and vasculitis panel in serum came negative. Serum and CSF AQP4-IgG and MOG-IgG were negative. After reviewing her MRI study, she was started on injection methylprednisolone 1 gram iv once a day for next 5 days. Clinically, patient’s mentation improved and returned to her baseline within next 3 days.
DISCUSSION
The International Consensus Diagnostic Criteria (ICDC) defines six core clinical characteristics of NMOSD: optic neuritis, acute myelitis, area postrema syndrome, acute brainstem syndrome, symptomatic narcolepsy or acute diencephalic clinical syndrome with NMOSD-typical diencephalic MRI lesions, and symptomatic cerebral syndrome with NMOSD-typical brain lesions. The criteria also introduced the possibility of a “seronegative NMOSD” to facilitate diagnosis in patients with unknown AQP4 IgG serostatus, if strict clinical and MRI requirements are met [2].With the absence of transverse myelitis and optic neuritis, this case demonstrates a very atypical presentation.
This patient’s chief complaint was negativism, mutism, waxy flexibility and stupor. Few reported cases have demonstrated symptoms of psychosis/catatonia in patients with NMOSD [7,8,9]. The MRI showed bilateral thalami and brainstem involvement, which could have contributed to her symptoms.
Apart from enhancement of the optic nerves, an MRI brain is normal at presentation in up to 84% of patients with NMOSD [10]. In this patient, bilateral thalamic and peri ependymal involvement was present on admission with clinical signs of catatonia.
A link between NMOSD with anti-AQP4 antibody positivity and autoimmune connective tissue disorders has been previously documented [11]. An array of autoimmune testing for conditions such as SLE, Sjogren’s syndrome, and rheumatoid arthritis were obtained in our patient and should be assessed in NMOSD patients.
Our patient presented with clinical diagnosis of catatonia with brain imaging suggestive of central nervous system demyelinating illness, more in favour of NMOSD. MRI brain showed diencephalon and peri ependymal involvement, areas rich in AQP4 channels. Though Anti-AQP4 IgG were not detectable in serum and CSF. MRI brain did not reveal any altered signal in optic pathway and her Visual Evoked Potential testing could not be done. The patient’s CSF came out to be negative for viral and bacterial infections. Her diagnosis of seronegative NMOSD could not be established according to ICDC, 2020 criteria as none of the 3 core clinical syndromes (optic neuritis, longitudinally extensive transverse myelitis, area postrema syndrome) were present in this patient. However, we kept seronegative NMOSD as diagnosis of this patient based on the following findings- age of presentation, involvement of AQP4 channel rich areas in brain as evident on MRI brain and excellent response to pulse methylprednisolone therapy.
NMOSD can present across a broad spectrum of clinical findings and is potentially debilitating. Therefore, in young female patients with catatonia as presentation and thalamic lesions, it is only likely that NMOSD should be high on our differential; with early use of high-dose pulse steroid therapy to prevent long-term disability.
CONCLUSION
We discuss a rare presentation of NMOSD as catatonia. Unique to this case is the extensive nature of the brainstem lesions, which would otherwise delay the diagnosis. Differentials include viral encephalitis, autoimmune encephalitis, metabolic disorders, and other infiltrative disorders. Treatment is with pulse steroid therapy, IVIG, plasmapheresis and immune modulation drugs.
CONFLICT OF INTEREST
None.
REFERENCES
- Jarius S &Wildemann B. The History of Neuromyelitis Optica. J Neuroinflammation. 2013;10(1):8.
- Wingerchuk, D., Banwell, B., Bennett, J., Cabre, P., Carroll, W., Chitnis, T., de Seze, J., Fujihara, K., Greenberg, B., Jacob, A., Jarius, S., Lana-Peixoto, M., Levy, M., Simon, J., Tenembaum, S., Traboulsee, A., Waters, P., Wellik, K. and Weinshenker, B., 2020. International Consensus Diagnostic Criteria For Neuromyelitis Optica Spectrum Disorders.
- Barnett Y, Sutton IJ, Ghadiri M et-al. Conventional and Advanced Imaging in Neuromyelitis Optica. AJNR Am J Neuroradiol. 2013;
- Sarbu N, Shih RY, Jones RV et-al. White Matter Diseases with Radiologic-Pathologic Correlation. Radiographics. 2016;36 (5): 1426-47.
- Fink M. Rediscovering catatonia: the biography of a treatable syndrome. Acta Psychiatrica Scandinavica. 2013 Jan;127:1-47.
- Remberk B, Szostakiewicz Ł, Kałwa A, Bogucka-Bonikowska A, Borowska A, Racicka E. What exactly is catatonia in children and adolescents. Psychiatr Pol. 2020 Aug 31;54(4):759-75.
- Ruiter AM, Meilof JF, Somanje-Bolweg RR, Van Gorsel E, Kalkers NF. Autonomic Dysregulation, Cognitive Impairment, and Symptoms of Psychosis as an Unusual Presentation in an Anti-Aquaporin 4-Positive Patient. Case Reports in Neurology. 2017;9(1):12-16.
- Alam A, Patel R, Locicero B, Rivera N: Neuromyelitis optica presenting with psychiatric symptoms and catatonia: a case report. Gen Hosp Psychiatry 2015;37:274.e1–2.
- Woolley J, Douglas VC, Cree BA: Neuromyelitis optica, psychiatric symptoms and primary polydipsia: a case report. Gen Hosp Psychiatry 2010;32:648.e5–8.
- Sellner J, Boggild M, Clanet M, et al. EFNS guidelines on diagnosis and management of neuromyelitis optica. European Journal of Neurology. 2010;17(8):1019-1032.
- Jarius S, Jacobi C, de Seze J, Zephir H, Paul F, Franciotta D, et al: Frequency and syndrome specificity of antibodies to aquaporin-4 in neurological patients with rheumatic disorders. Mult Scler2011;17:1067–1073.