Post Graduate Clinic; Approach to Cerebellar Ataxia: A Coherent Review
Anurag Rohatgi
Director Professor, Department of Medicine, Lady Hardinge Medical College, New Delhi
MacDonald Critchley in 1965 described that the so called silent areas of Brain: Cerebellum and Frontal lobe are the most vital areas of the Neuraxis which require a keen sense of observation. Cerebellar Ataxias are extensively encountered around the Globe and these heterogenous group of Neuro degenerative disorders have cumbersome lists of classification schemes. The word “Ataxia” is derived from a Greek word a’taxis’ meaning without order. Apart from Cerebellar disorders, possible sites of lesions for Ataxia include Large fibers of Peripheral nerves, Dorsal Root ganglion, Posterior Columns, Frontal Lobe with it’s connections and Vestibular system.
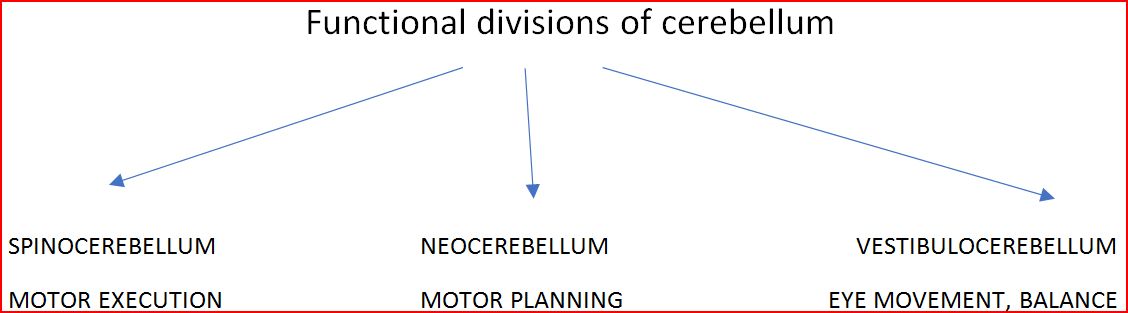
Cerebellar Ataxias manifest as irregularities in the Rate, Rhythm, Force and Amplitude of voluntary movement, mainly involving the Initiation and Termination of motion culminating in Dysynergia, Terminal tremors and Overshoot of limbs. Cerebellum influences the Ipsilateral side of the body. Anatomically cerebellum is subdivided into Anterior Lobe, Posterior Lobe and Floculonodular Lobe with a central Vermis. Vermis mainly controls the Gait and Stability of Head; Trunk. Cerebellar Hemispheres control Tone of Limbs, Eye movements, Speech and Coordination. So, apart from Limb Ataxia and Gait Ataxia Cerebellar lesions comprise of a spectrum of manifestations ranging from Dysarthria (Scanning and Staccato speech), Titubation , Intention tremors to Gaze evoked Nystagmus.
The basic classification scheme of Cerebellar ataxias comprises the division of Ataxias into two main groups that is: Hereditary Ataxias and Acquired ataxias. The salient features of differentiation of these two groups are provided in Table 1.

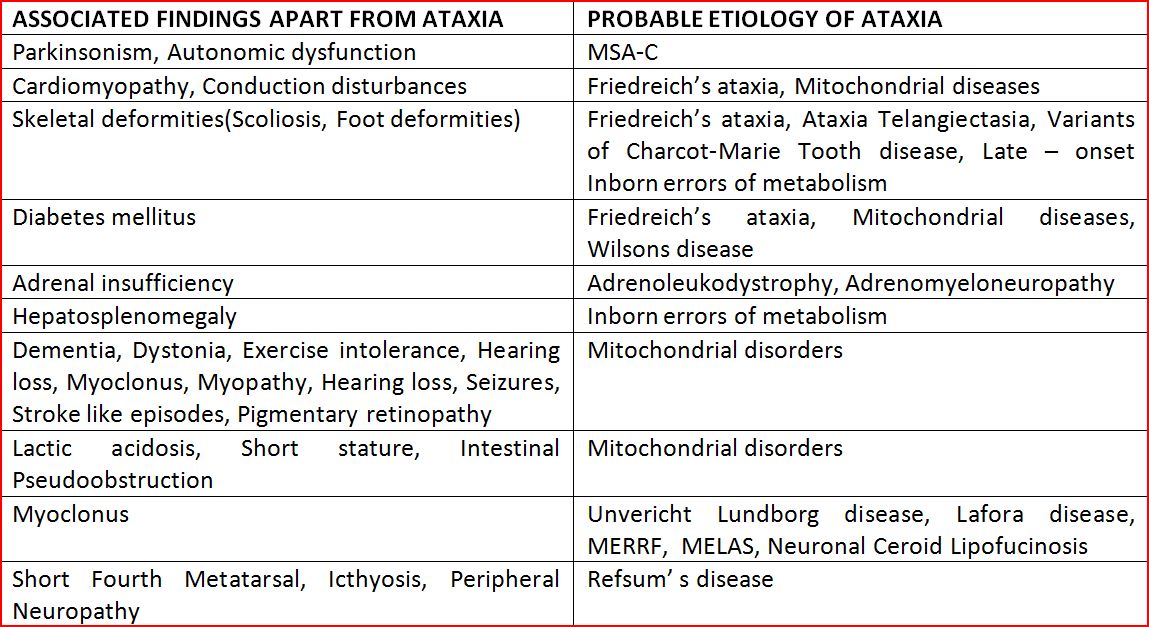
A pragmatic examination seeks to evaluate Clinical presentation in the form of pure Ataxic form or associated features of Ataxia “Plus”. The salient clinical examination findings in Ataxia plus syndromes that may provide clue to the diagnosis of Ataxia are given in Table 2.
Genetic Ataxias are further classified into:
Autosomal Dominant Cerebellar Ataxias,
Recessively inherited Ataxias,
Maternally inherited Ataxias
and Sporadic Ataxias.
Autosomal Dominant Ataxias have an average age of onset in the Third decade and most of these are indistinguishable from each other especially in the early stages except by Genetic testing. Most common type of Spinocerebellar Ataxia in the world is the Machado Joseph disease (SCA-3). Apart from SCA 6 and SCA 5 most of the other subtypes of SCA present with features other than Cerebellar Ataxia. Subtypes of Autosomal Dominant ataxias are given in Table 3.

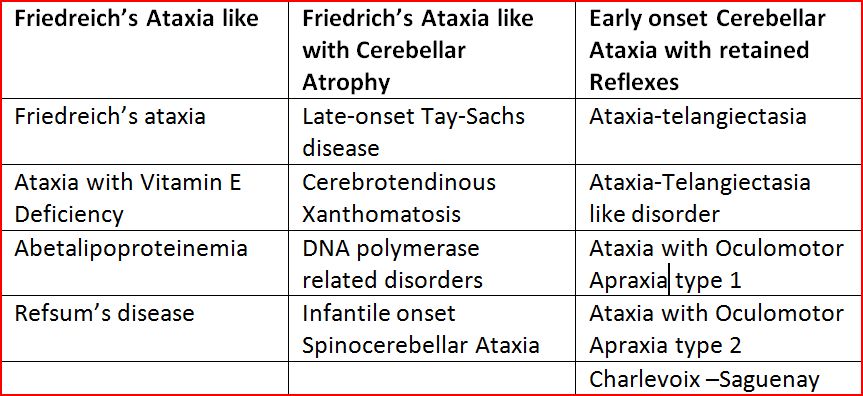
Recessive Ataxias most often have an onset before the age of 25 years. The most common Autosomal Recessively Inherited Ataxia is Friedreich’s Ataxia. Recessively inherited Ataxias can be phenotypically classified into three types as given in Table 4.
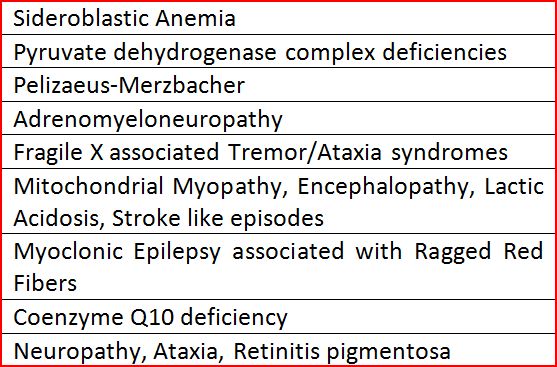
Maternally inherited Ataxias (Table 5) are suspected when the defective genetic material always has an origin from the Maternal side of a family. Mother may or may not be symptomatic. Both her Sons and Daughters have equal risk of developing symptoms, but in case of X linked disorders the Female carriers will not manifest symptoms.
In case of Sporadic Ataxias, Non genetic Ataxias are more common than Inherited Ataxias in a ratio of 2:1. In such cases we should obtain a detailed Family and Environmental history and should rule out all possible Acquired causes.
INDIAN SCENARIO
In the exams, one can also approach Ataxias as Acute, Sub Acute and Chronic Ataxias and keep these etiologies as the differentials in the Indian scenario as depicted in table 5.
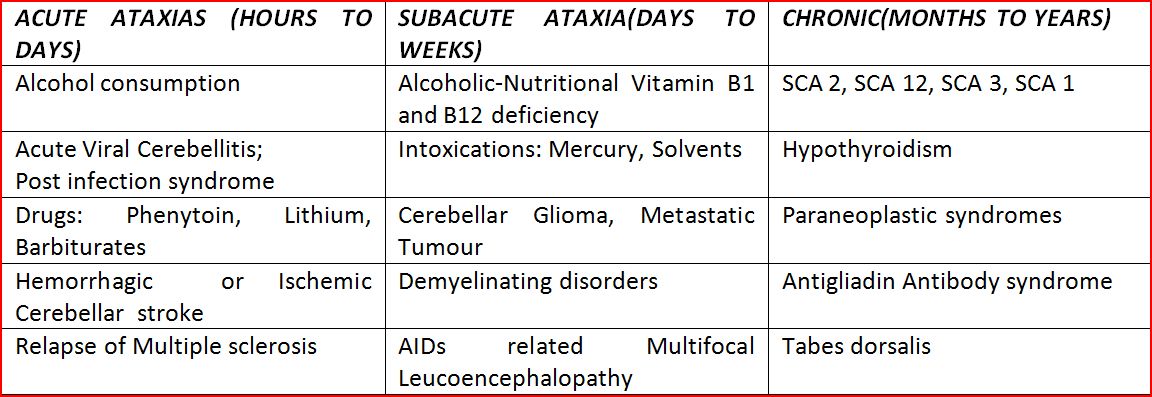
Acute onset Ataxia:
With no progression of the deficit suggests Injury, Stroke and Anoxia. Here Ataxia develops within a span of minutes to a few days. Similar presentation can occur with Toxins like Ethanol, Mercury, Thallium, Toluene; other solvents and Medications like Pheytoin, Carbamazepine, Phenobarbital and Lithium. Infectious processes like Viral Cerebellitis, Basilar meningitis and Cerebellar Abcess can also sometimes have an Acute presentation.
Subacute onset
With progression lasting weeks to months can suggest Infectious, Inflammatory, Immune or Neoplastic processes and Metabolic derangement . Superficial Siderosis usually following Trauma and SAH can also have a Sub acute presentation. Even though rare, Anti GAD Ataxia can also run a Subacute course and early recognition of this pathology helps us in initiating Immunotherapy as it is has a good reponse.
Chronic Course
Genetic ataxias should be thought about in a case of Chronic progressive Ataxia with a very slow protracted course. Even the absence of a clear Family history does not rule out the role of Genetic factors in an apparently Sporadic ataxia. This can be due to the fact that information wasn’t sought or the information was not available in cases of Non cooperation, loss of contact, Paternity issues, or Adoption of child. The existence of Non dominant Inheritance patterns or specific Genetic processes like Anticipation, Incomplete penetrance or Mosaicism can also explain the absence of a Family history in these subjects.
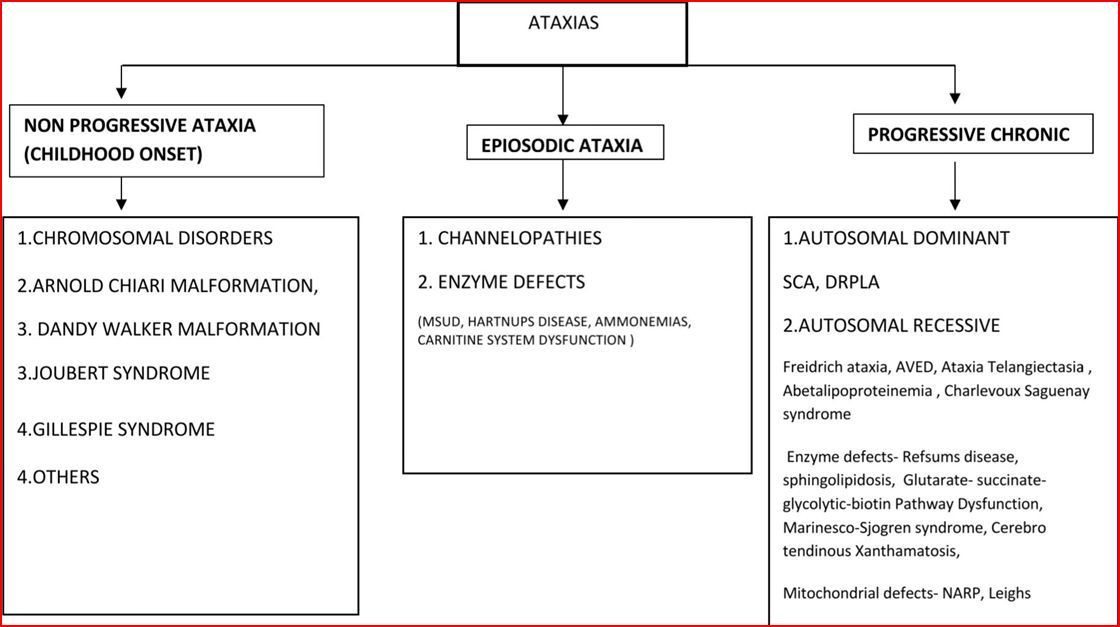
So a simplified approach can be adopted which encompasses Age of onset, Pattern of progression (Progressive, Non progressive and Episodic), Gender, Family tree diagram, Associated Neurological and Systemic features. A simplified Flow chart 2 given below presents an Approach to Pattern of progression of clinical course of Ataxic disorders.
“The Good Physician treats the Disease; the Great Physician treats patients who has the disease” Sir William Osler remarked. The way in which he lived his personal life conveyed to us that Clinicians should always have a Disciplined, Focussed and Scientific approach towards each symptoms, as it can always help to narrow down the differentials.